
Every year, 300,000 babies are born with hemoglobin disorders, and a major proportion of them have Sickle Cell Disease. In order to crack down on this congenital disorder, the World Health Organization (WHO) observes June 19 of every year as the ‘World Sickle Cell Day’, to educate people on this condition and increase access to healthcare services for patients with this disease.
What causes SCD
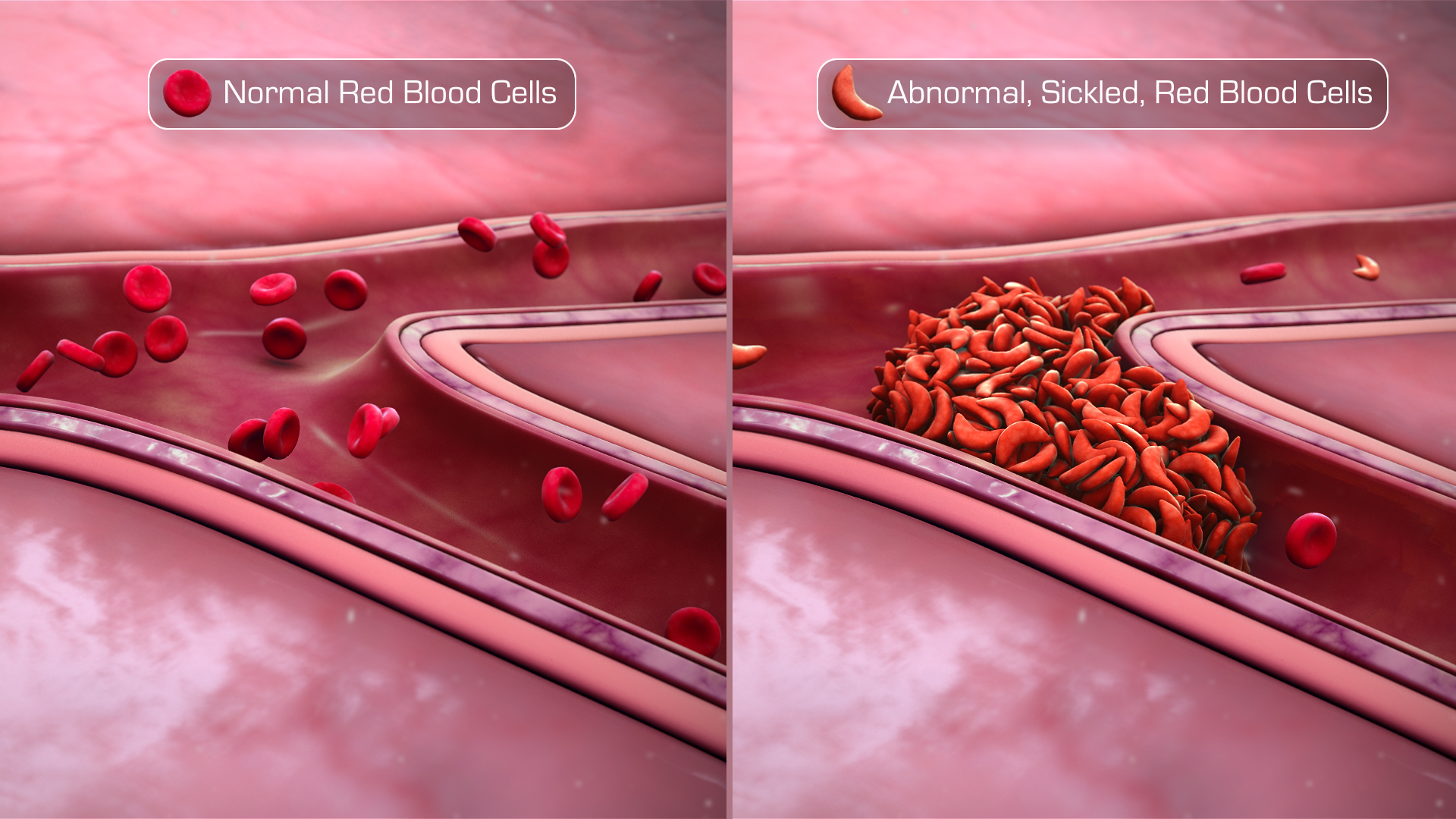
When two copies of a mutant form of the HBB gene are inherited, a series of genetic changes takes place in human body. A missense mutation, i.e. GTG to GAG, causes glutamic acid to be substituted with valine, thereby producing an abnormal structure of hemoglobin known as Hemoglobin S. RBCs containing Hemoglobin S are called as Sickle Cells because of their crescent-shaped pointy structure, unlike the biconcave structure of a normal RBC.
SCD is an autosomal recessive disease. Individuals with a recessive allele of the mutant gene (Hb S gene) can be carriers of the disease but a person carrying both the alleles has the disease.
Types of SCD
There are 4 types of SCDs depending on the genes inherited:
- When the progeny receives Hb S genes from both its parents, the disease is known as Hemoglobin SS type. Patients with this condition have the most fatal anemias.
- When the progeny receives the Hb S gene from one parent and Hb C gene from another, the disease is of Hemoglobin SC type. The symptoms are less severe than in Hemoglobin SS type.
- When the progeny receives the HBB gene along with the Hb S gene, the globin production reduces along with the size of the RBCs, but the biconcave structure remains. The condition is known as Hemoglobin S Beta Thalassemia.
- There is another severe form of beta thalassemia, known as Beta-zero Thalassemia. The symptoms are as severe as in Hemoglobin SS type and it’s complicated by nil production of globin.
How does SCD affect
Sickle cells inhibit RBCs from moving freely inside blood vessels, specially through narrow peripheral arteries, and thus clog the arteries and cause pain.
A baby born with sickle cell disease starts showing symptoms as early as at 4 months, when the fetal hemoglobin is replaced with Hemoglobin S and the cells become sickle-shaped.
SCD has fatal manifestations as the individual grows from infancy to an adult. Most common are anemia, excessive pain in the upper and lower extremities, delayed puberty, and infections, whereas the most fatal ones are acute chest syndrome, cerebrovascular accidents, and progressive vasculopathy, to name a few.
Children with SCD are prone to infections due to their impaired immune systems, and malfunctioning of lymphatic organs. Therefore, antibiotics have to be administered every now and then along with pain medications. SCD patients suffer with anemia because sickle cells have a short lifespan of only 10-20 days, compared to normal RBCs (110 - 120 days).
The shape distortion impairs the blood flow through the lumen of arteries and veins. Hemoglobin S induces intravascular hemolysis, which reduces the capacity of the cell to transport oxygen.
Treatment Methods
Therapeutic methods are available that aid in lengthening the patient’s lifespan and managing the symptoms, but there is no cure yet for this genetic disorder. In order to survive, the patient has to go through periodic blood transfusions and medication throughout his life.
Since blood transfusions increase the iron content in the body, a new therapeutic procedure -- iron chelating therapy -- has been designed to overcome the transfusional iron overload. Hydroxyurea is another therapeutic agent given to SCD patients with severe anemia, so that chest pain and respiratory symptoms are kept in control.
Patients experiencing vascular pain, in the hands and feet, are advised to consume water excessively to keep the blood cells mobile. Bone marrow transplants are administered in cases of severe anemia and respiratory difficulties.
The severity of SCD is governed by the degree to which Hemoglobin S polymerizes in the cell and induces shape distortion. In the prenatal stages, this polymerisation is inhibited by fetal hemoglobin, but as age progresses exposure to oxygen polymerises Hb S and gives rise to ‘cell sickling’.
Preventing SCD
Detecting the sickle cell trait before conceiving a baby is the only prevention measure. Blood samples are tested for Sickle cell disease and thalassemia by Hemoglobin Electrophoresis. Tropical countries such as India, Zimbabwe, Nigeria and Mediterranean countries have been noted to have high-risk populations.
Things Everyone Must Know About Blood Donation
We all have one of the four common blood types: A, B, AB, and O, as categorized under the Karl Landsteiner’s blood grouping system. It’s interesting to note that while human blood has common components, like RBCs, WBCs, Plasma, and Platelets, Read More..






